Рак щитовидной железы

Рак щитовидной железы (РЩЖ) – самая частая опухоль эндокринной системы. Доля случаев РЩЖ среди злокачественных новообразований всех локализаций составляет около 1 — 1,5%. РЩЖ проявляется как структурная (узловая) патология щитовидной железы (ЩЖ). Распространенность узловой патологии ЩЖ в популяции чрезвычайно высока, узлы ЩЖ пальпаторно определяются у 4-7% населения, при УЗ-скрининге узлы в ЩЖ обнаруживаются в 50% случаев. В необлученной популяции частота карцином ЩЖ в структуре узловой патологии органа составляет 5%. Имеются сообщения о том, что в популяции, подвергшейся лучевому воздействию на ЩЖ, частота злокачественных опухолей в структуре узловой патологии ЩЖ существенно возрастает. Наиболее высок риск развития РЩЖ после облучения ЩЖ в детском и подростковом возрасте.
В последние три десятилетия отмечается неуклонный рост заболеваемости РЩЖ во всем мире. Данная тенденция прослеживается на всех континентах, за исключением Африки, что, вероятно, обусловлено низких уровнем диагностики. В Италии РЩЖ вторая по частоте злокачественная опухоль среди женщин до 45 лет. И только в Норвегии и Швеции заболеваемость РЩЖ имеет тенденцию к снижению. С 2000 по 2013 гг. заболеваемость РЩЖ в РФ выросла более чем в два раза и в 2015 году составила 8.5 случая на 100 тыс. населения в год. По темпу прироста РЩЖ занимает первое место среди злокачественных опухолей, варьируя в последние 10 лет (с 2006 по 2016гг), заболеваемость увеличилась на 29,53 %. Так же необходимо отметить, что РЩЖ — это заболевание, которое поражает людей трудоспособного возраста с пиком заболеваемости, приходящийся на возрастную группу 50 – 60 лет, что делает данную проблему социально значимой. Увеличение заболеваемости РЩЖ происходит практически только за счет увеличения количества выявленных случаев папиллярной карциномы (ПК), при сохранении относительно стабильного уровня заболеваемости медуллярным, фолликулярным и анапластическим РЩЖ. Рост заболеваемости РЩЖ регистрируется во всех экономически развитых странах мира и в последние 2-3 десятилетия ежегодный прирост в различных странах варьирует от 3 до 10%, причем повсеместно РЩЖ лидирует в темпах прироста среди злокачественных новообразований всех локализаций. Смертность больных РЩЖ составляет в среднем 0,5 смертельных исходов на 100 тысяч населения в год. В исследовании 2017 г из Кореи, при анализе смертности от рака ЩЖ за последние 20 лет были получены интересные данные: отмечался рост смертности от РЩЖ до 2004г, с 2005 до 2015г выявлено ежегодная тенденция к снижению данного показателя. Среди возможных причин снижения смертности авторы указывают раннюю диагностику РЩЖ, изменение стратификации факторов риска и стандартизированный подход к диагностике и лечению.
В исследовании 2017 года (Topstad D. Et al.), исследователи проанализировали изменения распространенности РЩЖ за период с 1970 по 2012г в Канаде и пришли к выводу, что быстрое увеличение частоты РЩЖ с начала 1990 года связано с «эпидемией» гипердиагностики клинически незначимых РЩЖ. Данный вывод авторы делают, основываясь на том, что смертность от РЩЖ осталась без динамики за данный период, и не увеличилось число выявляемых агрессивных случаев рака ЩЖ. Увеличение количества впервые выявленных случаев РЩЖ связано не столько с истинным ростом заболеваемости РЩЖ, сколько с улучшением его выявляемости, свидетельством чего, по мнению ряда авторов, является увеличение случаев РЩЖ исключительно за счет папиллярного гистологического типа, и преимущественно микрокарцином.
Следует отметить, что, несмотря на значительное увеличение количества выявляемых микрокарцином, также растет и количество впервые выявленных опухолей большего размера.
В большинстве случаев (90-95%) РЩЖ возникает из фолликулярных эпителиальных клеток. Данный тип рака гистологически подразделяется на папиллярную (ПК), фолликулярную (ФК), включая так называемую Hurthle-клеточную карциному, низкодифференцированную (НДК) и анапластическую (АПК) карциномы. Около 3% случаев РЩЖ приходится на медулярную карциному, возникающую из парафолликулярных С-клеток. Тяжелым последствием эволюции папиллярного и фолликулярного рака является конверсия высоко дифференцированной опухоли (ВДРЩЖ) в низкодифференцированную и анапластическую карциному, хотя редко данные типы новообразований могут возникать de novo.

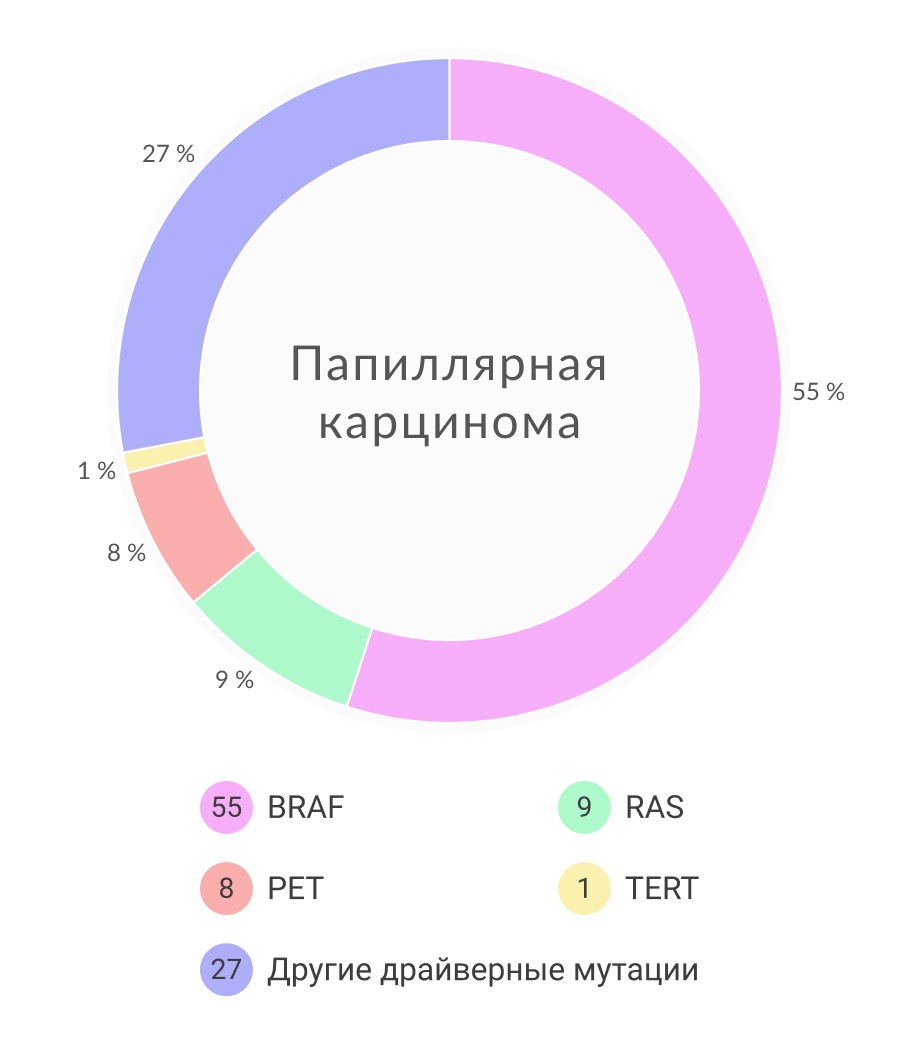
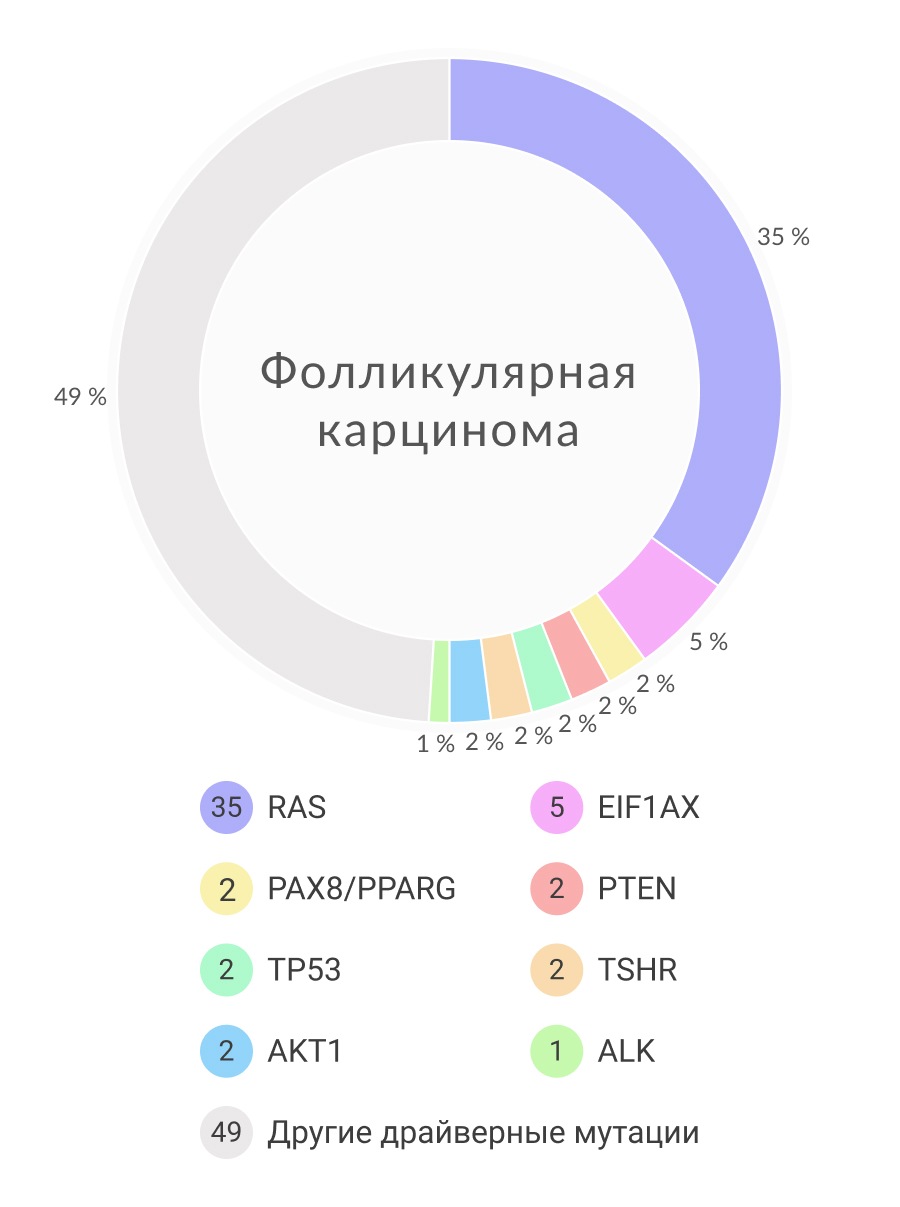
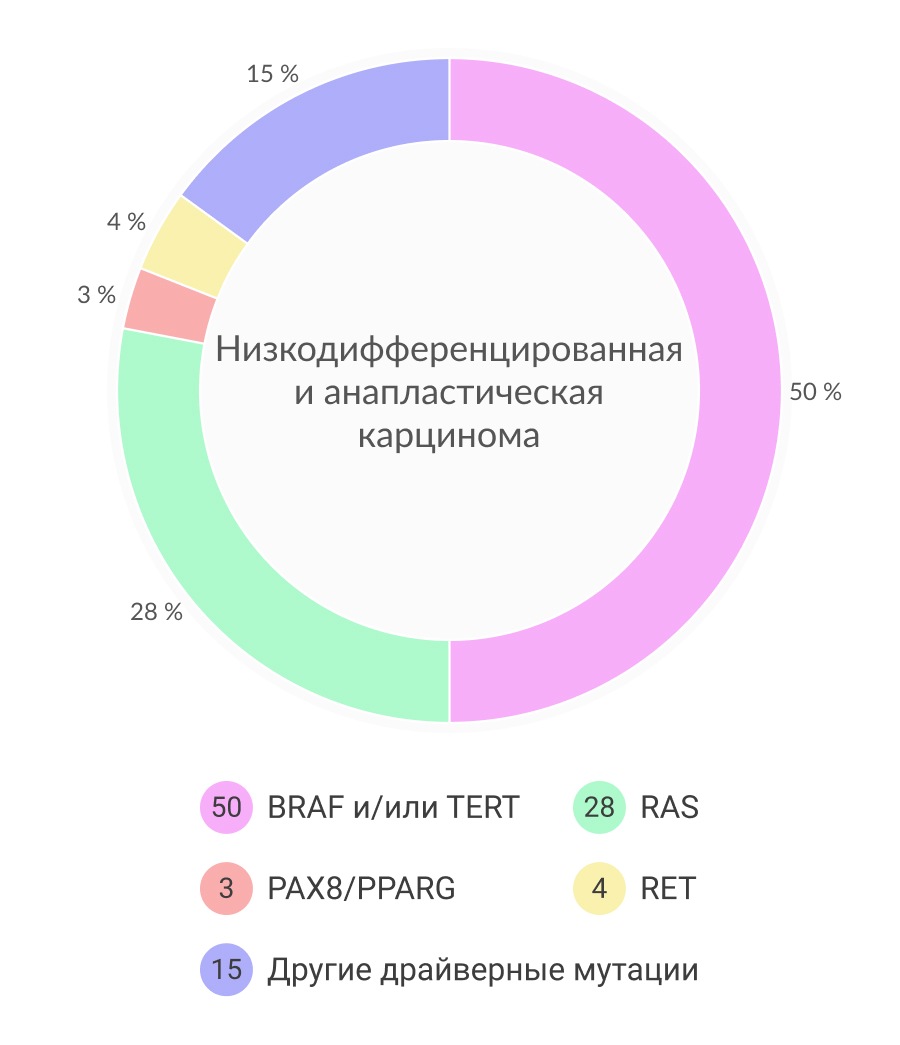
Наиболее часто встречающийся гистологический тип злокачественной опухоли — ПК, доля которого составляет в среднем 80-85% всех случаев РЩЖ. Этот гистологический тип РЩЖ преобладает в детской и подростковой возрастной группе (92-96% случаев), у взрослых пациентов до 65 лет его доля составляет около 80% случаев, в старшей возрастной группе — 70-75% случаев. ФК диагностируется примерно у 5-20% заболевших, у 5-10% больных выявляется медуллярный РЩЖ. Низкодифференцированный РЩЖ (НДК) встречается в 0,5-3 % случаев. Анапластический РЩЖ (АПК, недифференцированный РЩЖ) диагностируется в 0,1- 1% случаев. Другие гистологические типы злокачественных опухолей ЩЖ, такие как плоскоклеточный рак и лимфома, встречаются исключительно редко и рассматриваются как казуистика.
Узлы щитовидной железы (УЩЖ) выявляются в ходе ультразвукового исследования у 60% — 70 % населения в возрасте старше 50 лет. Большинство УЩЖ представляют собой доброкачественные узловые образования, которые чаще всего не требуют хирургического лечения, однако в 5 % — 10 % случаев под «маской» существующего узлового зоба протекает злокачественный процесс.
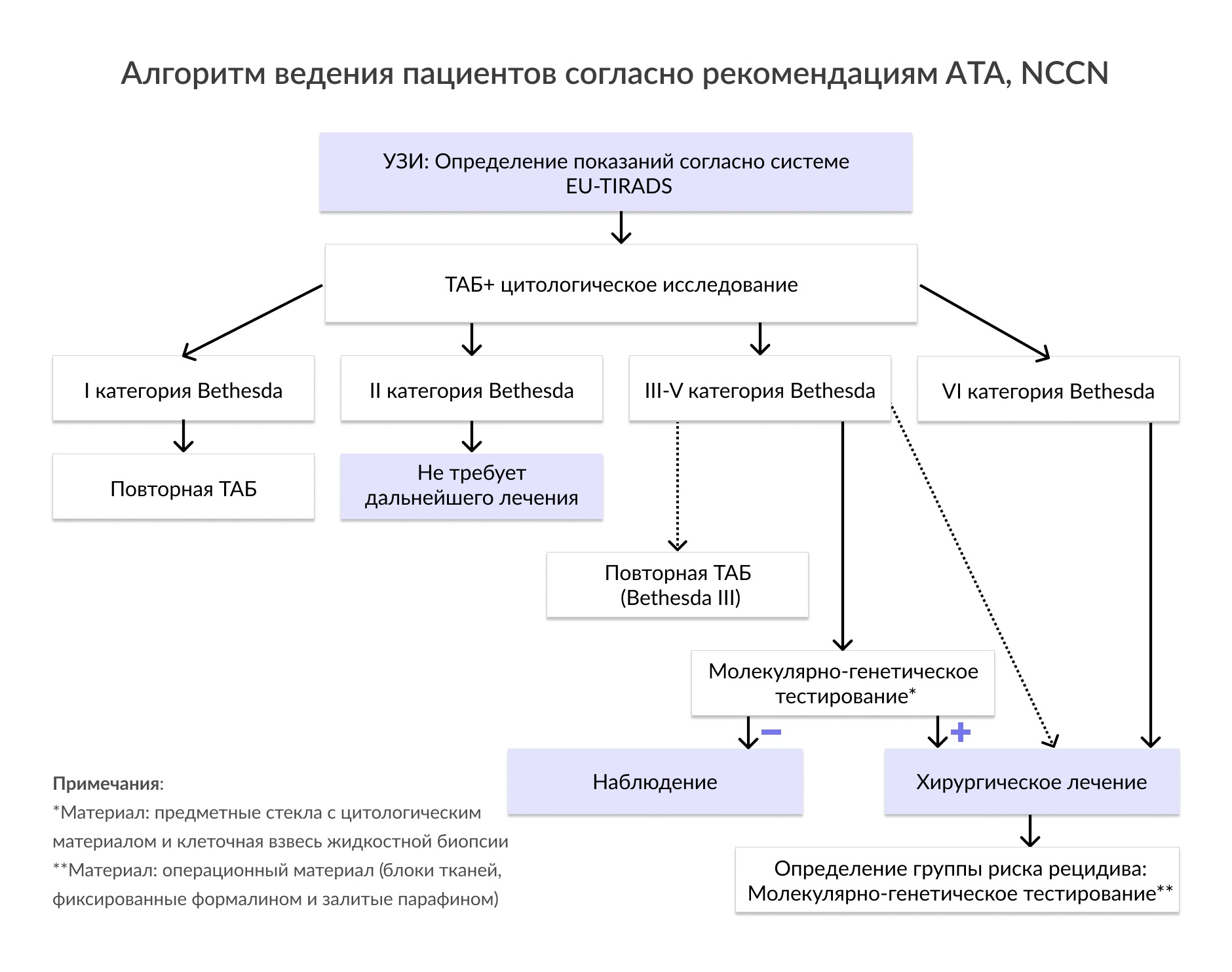
Наиболее важным с клинической точки зрения является дифференциальная диагностика развивающегося злокачественного процесса и доброкачественной пролиферации. Единственным способом прямой оценки морфологических изменений в ЩЖ в настоящее времени является ее биопсия. Введение в практику биопсии ЩЖ позволило сократить количество операций на ЩЖ на 35-75%, в то же время, выявляемость РЩЖ увеличилась вдвое. Из двух известных вариантов метода – тонкоигольной аспирационной биопсии (ТАБ) и трепанобиопсии. Именно ТАБ с последующим цитологическим исследованием является золотым стандартом диагностики РЩЖ. На сегодняшний момент, в соответствии с системой Bethesda, цитологическое заключение включает в себя несколько диагностических категорий: недиагностический или неудовлетворительный пунктат (Bethesda I), доброкачественный (Bethesda II), атипия неопределенного значения или фолликулярные изменения неопределенного значения (Bethesda III), фолликулярная неоплазия или подозрение на фолликулярную неоплазию (Bethesda IV), подозрение на рак (Bethesda V), рак (Bethesda VI). Несмотря на комплексность системы Bethesda, данный метод имеет ряд существенных недостатков. Одним из них является возможность получения цитологии неопределенной значимости, когда специалист не может достоверно подтвердить или исключить диагноз РЩЖ ввиду отсутствия в некоторых опухолях характерных признаков. Такие образцы относят к III-V цитологическим категориям по классификации Bethesda. Пациентам с цитологическим заключением Bethesda III-V проводят повторную процедуру ТАБ или диагностическую операцию, обычно лобэктомию, которую можно избежать для большинства пациентов с доброкачественными новообразованиями. В то же время, оставшимся пациентам, узлы которых имели неясную клиническую значимость, но оказались злокачественными по результатам постоперационного гистологического исследования, требовалась повторная операция с полной тиреоидэктомией.
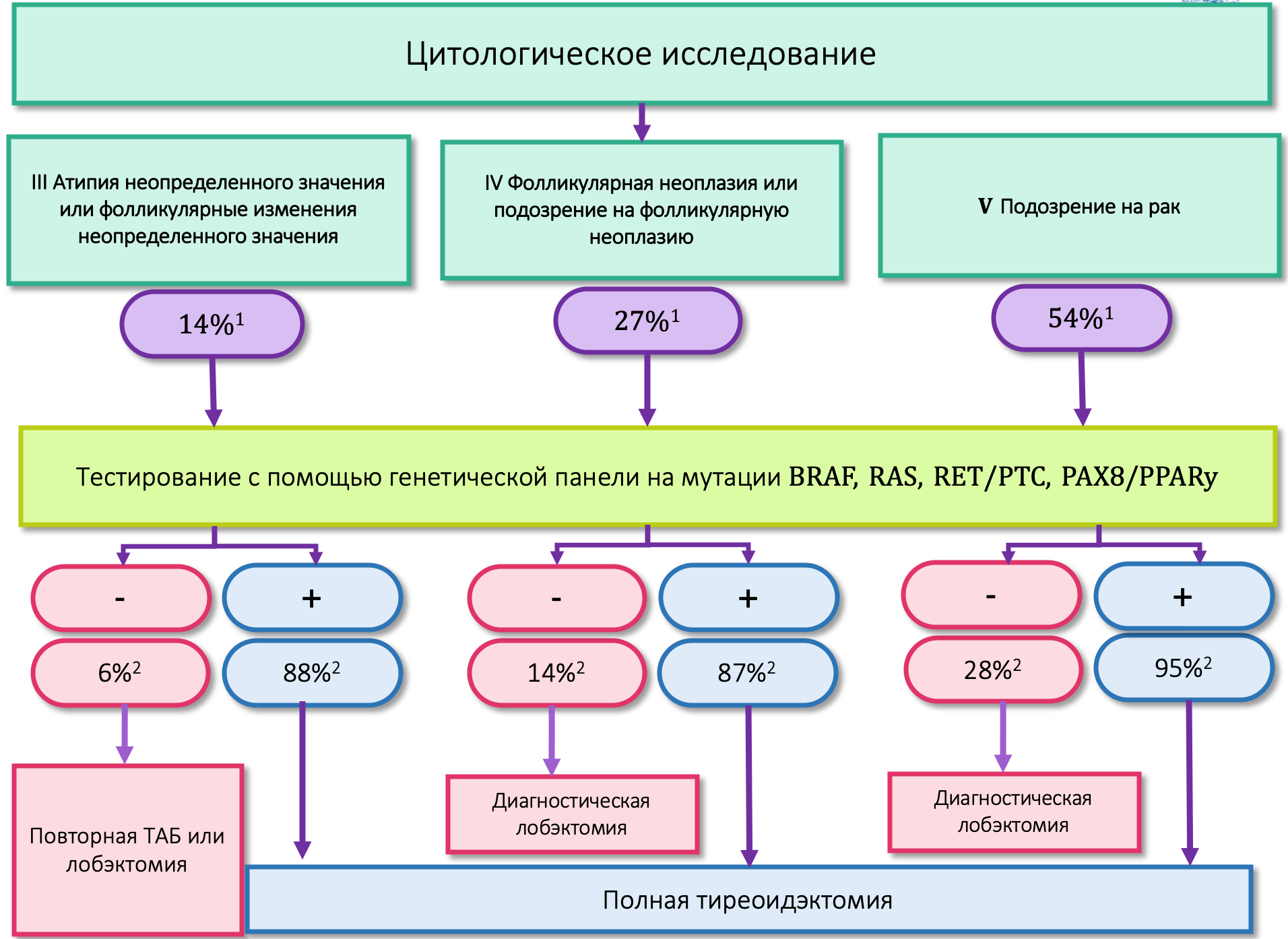
Использование дополнительных молекулярно-генетических методов диагностики в некоторых случаях позволиляет избежать как ненужной операции, так и повторного вмешательства. В связи с этим в различных международных рекомендациях (NCCN, ATA, ESMO) для пациентов с неясной цитологией показано проведение генетического анализа на широкий спектр аберраций в генах BRAF, TERT, KRAS, NRAS, HRAS, RET/PTC, PAX8/PPARG, который позволяет увеличить выявляемость РЩЖ и сократить время постановки диагноза.
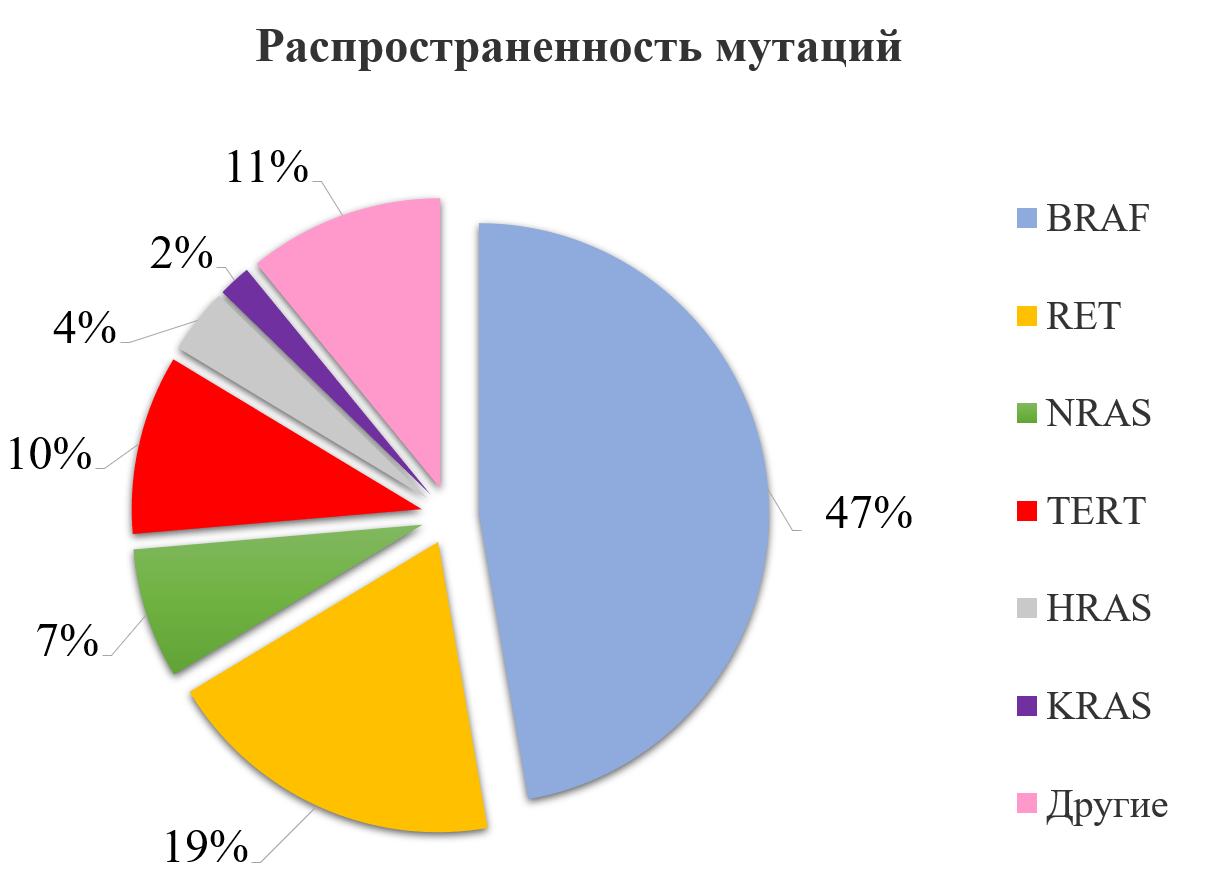
Большинство мутаций при РЩЖ возникает в генах, кодирующих белки MAPK-киназного и PI3K–AKT путей. MAPK-протеинкиназы — группа белков, включающая три семейства протеинкиназ — p38, JNK/SAPK (c-Jun N-terminal kinase/ Stress-activated protein kinase) и ERK (extracellular signal-regulated kinase). Практически во всех случаях активация протеинкиназ семейства ERK связана с клеточным выживанием и стимуляцией пролиферации, а активация протеинкиназ семейства p38 и JNK – с индукцией апоптоза. Поскольку в ответ на различные клеточные стимулы в клетках должна меняться экспрессия тех или иных генов, основными мишенями MAPK-протеинкиназ являются траскрипционные факторы. Несмотря на то, что транскрипционные факторы, локализованные в ядре, являются важными субстратами MAPK, только часть активированных в цитоплазме MAPK транслоцируется в ядро. Немалая часть MAPK остается в цитоплазме и других субклеточных компартментах, где роль этих протеинкиназ изучена гораздо меньше. Известно, что MAPK могут регулировать экспрессию генов и на посттранскрипционном уровне, используя в качестве субстратов цитоплазматические белки. Активация MAPK является одним из самых важных этапов канцерогенеза РЩЖ. Гены, аберрации в которых обнаруживаются при РЩЖ, кодируют тирозин-киназные трансмембранные белки RET, NTRK1, а также внутриклеточные вторичные посредники сигнала BRAF и KRAS. Аберрации в данных генах являются взаимоисключающими, обнаруживаются в 70% случаев папиллярной карциномы и ассоциированы с характерными гистопатологическими и биологическими характеристиками. Гистологически BRAF-ассоциированный РЩЖ представлен высококлеточным и классическим вариантами папиллярного рака, экстратиреоидным распространением, склонностью к дедифференцировке. В то же время RAS-ассоциированный рак характеризуется фолликулярным вариантом ПК (ФВПК) и частой инкапсуляцией.
RET/PTC
Транслокация RET/PTC встречается в основном при папиллярном РЩЖ. В результате транслокации ген RET сливается с рядом других генов. Получающиеся химерные конструкции включают в себя часть гена RET, кодирующего тирозинкиназный домен рецептора RET, и активный промотор другого гена, который осуществляет экспрессию и лиганд-независимую димеризацию протеинового комплекса RET/PTC. Данная химерная конструкция ведет к постоянной стимуляции MAPK-сигнального пути и злокачественного перерождения клеток. Наиболее частыми перестройками являются RET/PTC1 и RET/PTC3, в которых RET соединён с CCDC6 (H4) и NCOA4 (ELE1 или RFG) соответственно. Обе перестройки представляют собой парацентрические, интрахромосомные инверсии, так как все гены, образующие структуру, находятся на одной и той же 10 хромосоме. С другой стороны, RET/PTC2 и 9 других ранее описанных RET/PTC перестроек представляют собой интерхромосомные конструкции, формирующиеся при слиянии части гена RET с генами, расположенными на других хромосомах. Распространенность и специфичность RET/PTC перестройки при папиллярном РЩЖ варьирует в различных исследованиях. Частично данные вариации объясняются реальной разницей распространённости описанной аберрации в различных возрастных группах и наличием пациентов, перенесших ионизирующее облучение. Кроме этого, гетерогенность объясняется неравномерным распределением мутации в опухоли и различающейся чувствительностью используемых методик. В некоторых случаях перестройка RET/PTC является клональной и обнаруживается в большинстве клеток опухоли. Также аберрация RET/PTC может быть неклональной и обнаруживаться только в небольшом количестве клеток высокочувствительными методами ДНК диагностики. Клональная RET/PTC-перестройка обнаруживается в 10-20% случаев папиллярного рака и является специфическим маркером данного типа РЩЖ. В то же время неклональная перестройка RET/PTC обнаруживается намного чаще как при папиллярном раке, так и в других типах РЩЖ и доброкачественных образованиях (до 10-45% доброкачественных узлов и образований имели неклональную мутацию RET/PTC). Другая хромосомная перестройка, встречающаяся при папиллярном РЩЖ, затрагивает ген NTRK1, но встречается намного реже, чем перестройка RET/PTC. Ген NTRK1 располагается на хромосоме 1q22 и может сливаться как минимум с тремя различными генами, располагающимися на той же или других хромосомах. Перестройка NTRK1 наблюдалась в 10-15% случаев папиллярного РЩЖ в одном из исследований, хотя, вероятнее всего, распространенность данной аберрации не превышает 5%.
RAS
Гены HRAS, KRAS, NRAS кодируют G-белки, располагающиеся на внутренней части клеточной мембраны и переносящие сигналы, возникающие с мембранных тирозинкиназных рецепторов и G-связанных рецепторов по сигнальным путям MAPK и PI3K–AKT. Активирующие мутации в генах RAS чаще всего находятся в 12, 13 и 61 кодонах. При РЩЖ мутации в кодонах 61 генов NRAS и HRAS встречаются чаще всего. Мутации в генах RAS обнаруживаются в 10-20% случаев ПК, 40-50% случаев ФК и 20-40% случаев НДК и АПК. Среди ПК, практически все RAS-ассоциированные опухоли формируют неопластические фолликулы без папиллярных структур и, таким образом, диагностируются как ФВПК. Мутации в генах RAS встречаются в 20-40% доброкачественных фолликулярных аденомах. Обнаружение мутаций в RAS-генах в доброкачественных аденомах говорит о том, что RAS-положительные фолликулярные аденомы могут служить прекурсорами для RAS-положительных фолликулярных карцином и ФВПК. Более того, RAS мутации предрасполагают высокодифференцированный рак трансформироваться в анапластические образования.
BRAF
BRAF представляет собой серин-треониновую киназу, которая переносится к клеточной мембране после связывания и активации RAS, что приводит к фосфорилированию и активации MAPK- киназы и связанных с ней вторичных посредников. При РЩЖ BRAF активируется точечными мутациями, малыми делециями или инсерциями и хромосомными перестройками. Наиболее частым механизмом активации является замена тимина на аденин в нуклеотидной позиции 1799, что приводит к замене валина на глутамин в позиции 600 транслируемого белка (Val600Glu).
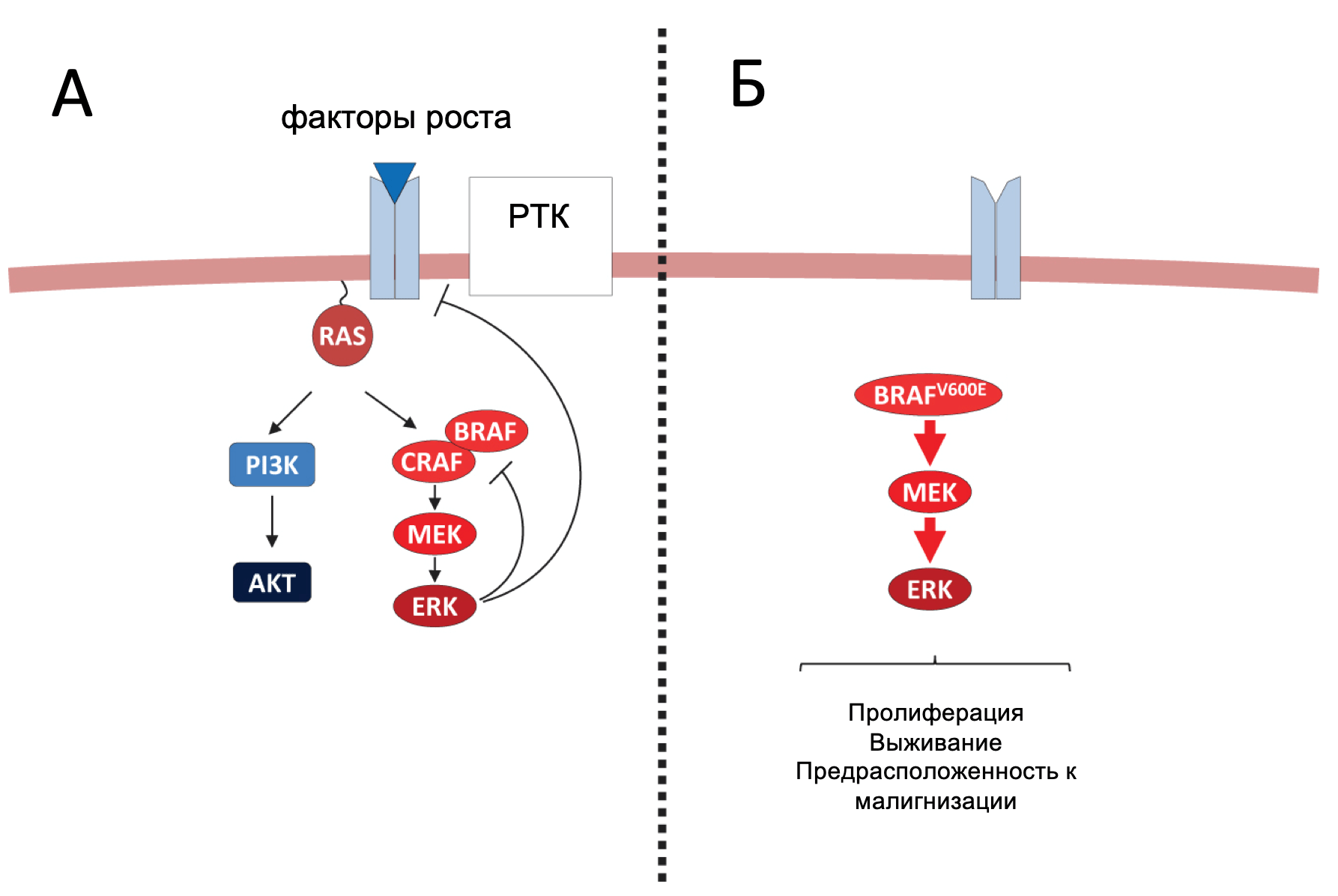
Данная аберрация встречается в 98-99% процентов всех мутаций, выявляемых в гене BRAF при РЩЖ. Другими менее распространенными генетическими изменениями являются Lys601Glu замена, инсерции и делеции около кодона 600, а также AKAP9/BRAF-перестройка. AKAP9/BRAF-перестройка представляет собой парацентрическую инверсию хромосомы 7q, что ведет к слиянию части BRAF-гена, кодирующей протеинкиназный домен и AKAP9-ген. Все точечные мутации и перестройки ведут к активации BRAF-киназы и хронической стимуляции MAPK-пути.
Мутация BRAF V600E представляет собой наиболее частую аберрацию, обнаруживаемую при папиллярном РЩЖ (40-50% случаев). Также данная мутация встречается в 20-40% случаев НДК и 30-40% случаев АПК. Гистологически во многих случаях BRAF-ассоциированных карцином обнаруживаются области высокодифференцированного папиллярного рака и BRAF-положительные клеточные линии присутствуют во всех областях опухоли, что говорит о том, что появление Val600Glu является ранним событием и предрасполагает к дедифференцировке опухоли. При папиллярном РЩЖ BRAF V600E-мутация обнаруживается чаще всего в двух вариантах ПК: классическом и высококлеточном. В то же время, BRAF Val601Glu гистологически может быть представлена фолликулярным вариантом.
PAX8-PPARγ
PPARγ представляет собой ядерный рецептор, участвующий в регуляции клеточного цикла и апоптоза. PAX8 в свою очередь является фактором транскрипции, регулирующий активность тиреоидо-специфических генов. PAX8/PPARγ-перестройка наблюдается при слиянии части гена PAX8, который кодирует фактор транскрипции, и гена PPARγ. Образование данной конструкции ведет к гиперэкспрессии химерного белка PAX8/PPARγ. Аберрация PAX8/PPARγ представляет собой прототипную мутацию, обнаруживаемую при фолликулярном РЩЖ, где она возникает в 30-35% случаев. Данная перестройка редко встречается при фолликулярных аденомах (2-13%) и ФВПК (1-5%). Мутация PAX8/PPARγ ассоциируется с развитием фолликулярной карциномы в раннем возрасте и высоким уровнем сосудистой инвазии. С точки зрения клинической значимости, обнаружение мутации PAX8/PPARγ говорит о фолликулярной структуре опухоли с предрасположенности к ранней сосудистой инвазии, но в то же время данная аберрация может встречаться и в доброкачественных образованиях. Несмотря на это, обнаружение перестройки PAX8/PPARγ при фолликулярной неоплазии предсказывает конверсию доброкачественного образования в рак.
Корреляция генотипа и фенотипа
Существует строгая корреляция между хромосомными соматическими аномалиями, характерными для ПК, и случаями облучения ионизирующим излучением. Перестройка RET/PTC обнаруживается у 80% пациентов с ПК, которые ранее подверглись воздействию радиации в терапевтических целях или случайно. Чернобыльская авария значительно увеличила частоту RET/PTC3 и новых RET/PTC-ассоциированных видов РЩЖ. Перестройка BRAF/AKAP9 также чаще всего встречается у пациентов с ПК, подвергнувшихся радиационному облучению. Нужно отметить, что точечные мутации в генах BRAF и KRAS редко встречаются в случаях РЩЖ, индуцированных излучением. Хотя точные механизмы формирования хромосомных перестроек при радиационном излучении не до конца понятны, считается, что ядерная и хромосомная архитектура вносит существенный вклад в перестройки RET/PTC и TRK, располагая склонные к рекомбинации участки генома в непосредственной близости друг к другу. Пространственная близость предрасполагает соседние гены к неверному ресоединению двуцепочечных разрывов, вызванных радиацией. Дополнительно индуцирование RET/PTC-перестройки может быть ассоциировано с хромосомной ломкостью. Регионы 10q11.2 и 10q21 расположения генов RET и CCDC6 соответственно, а также их партнеров при перестройках RET/PTC1 содержат два сайта ломкости: FRA10G и FRA10C. На культуре клеток было показано, что индукция ломкости в этих сайтах вызывало появление перестроек RET/PTC1. Хромосомная ломкость может вызываться или усиливаться гипоксией, этанолом, кофеином и другими эндогенными и экзогенными факторами. Таким образом, перестройка RET/PTC1 может вызываться и другими помимо радиации факторами. Данный мультифакториальный механизм, вероятнее всего, характерен для молодых пациентов с ПК.
В отличие от хромосомным перестроек, появление точечных соматических мутаций при РЩЖ чаще всего не связано с воздействием радиации. В китайском исследовании с выборкой более 1000 случаев ПК было показано, что появление BRAF V600E-мутаций ассоциировано с высоким уровнем потребления йода (OR=1,97). Результаты данного исследования не позволяют подтвердить причинно-следственную связь между высоким потреблением йода и мутагенезом. Но в случае подтверждения эти данные дадут биологический базис для понимания причин повышенной распространённости ПК по сравнению с фолликулярным в районах с повышенным потреблением йода. Другое исследование показало значительно большую встречаемость BRAF-позитивного папиллярного РЩЖ в вулканическом районе Сицилии, в воде которого наблюдаются повышенные концентрации бора, железа, ванадия, магния и других химических элементов. Эти данные позволили предположить, что индуцирование точечных мутаций BRAF может быть ассоциировано с чрезмерным воздействием различных химических элементов и соединений.
Мутации в генах BRAF и KRAS часто обнаруживают как в ВДРЩЖ, так и НДК, и АПК, что, вероятнее всего, говорит о том, что данные аберрации возникают на ранних этапах канцерогенеза. Часто при АПК и НДК обнаруживаются дополнительные мутации, которые не встречаются в случае дифференцированных форм РЩЖ. Появление новых аберраций на поздних этапах канцерогенеза может быть причиной начала дедифференцировки опухоли. Мутации в генах TP53, CTNNB1, а также мутации в генах, кодирующих белки PI3K–AKT сигнального пути, чаще всего обнаруживаются на поздних этапах эволюции опухоли. Точечные мутации гена TP53, кодирующего регулятор клеточного цикла p53, обнаруживаются в 50-80% случаев анапластической карциномы. Данные мутации менее часто обнаруживаются в НДК и редко в ВДРЩЖ. Мутации в гене TP53 ведут к потере функциональной активности важного онкосупрессора р53. Другим часто геном, подвергающийся альтерациям, на поздних стадиях канцерогенеза является CTNNB1, который кодирует β‑катенин, который участвует в адгезии и Wnt передачи сигнала. Точечные мутации в экзоне 3 встречаются в 60% случаев АПК и, реже, при НДК. Дедифференцировка РЩЖ также включает в себя прогрессивное накопление других мутаций, особенно в тех генах, которые кодируют белки PI3K–AKT сигнального пути. Так, например, 10-20% АПК и НДК имеют мутации в PIK3CA, 5–15% — в PTEN и 5–10% — в AKT1 генах.
За последние несколько лет были описаны новые соматические аберрации в промоторе гена TERT, обнаруживаемые при опухолях центральной нервной системы, мочевого пузыря, печени, меланомы и щитовидной железы.
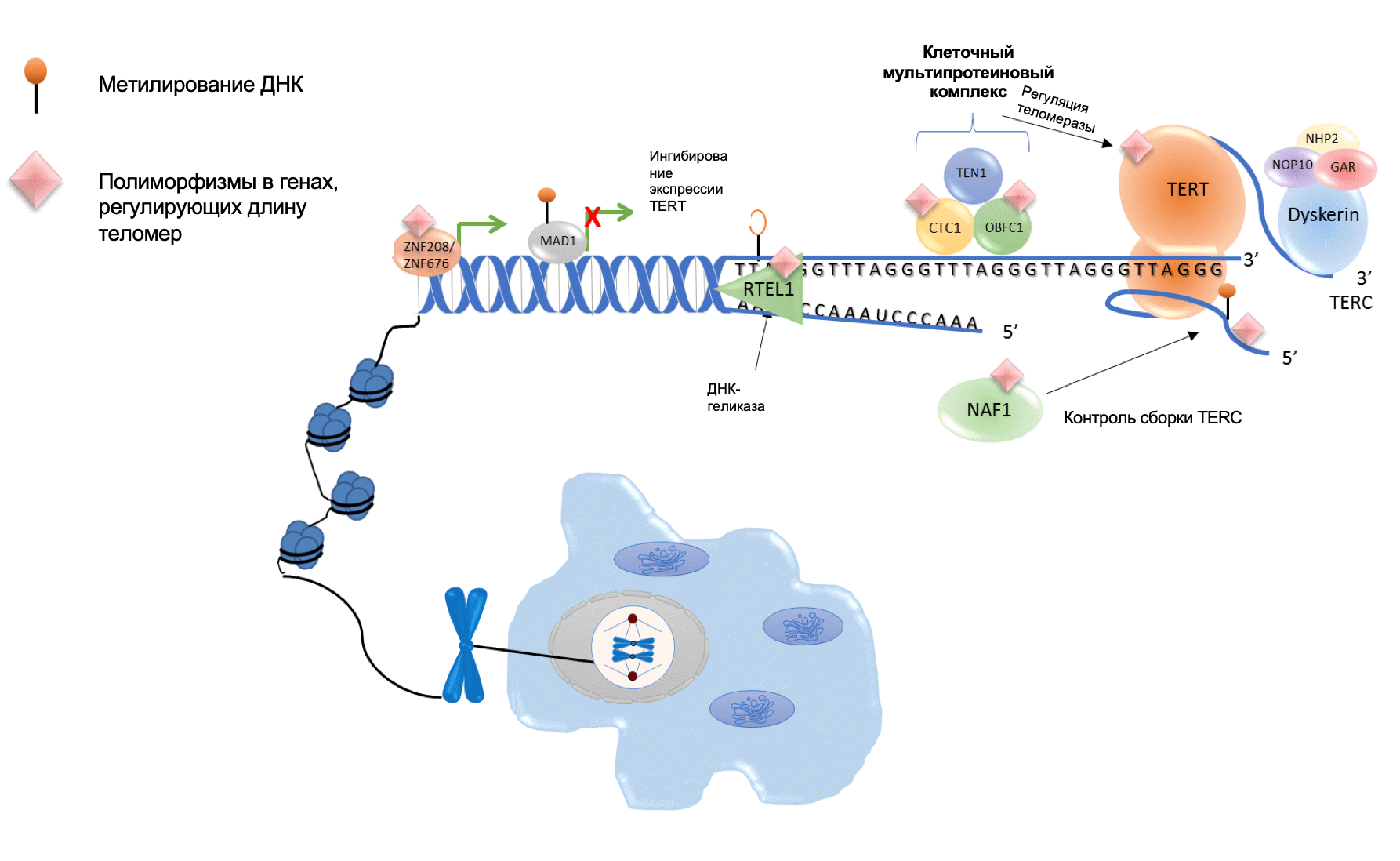
Теломераза представляет собой фермент, участвующий в элонгации теломер – комплекса нуклеопротеинов, располагающихся на конце хромосом, состоящих из TTAGGG-повторов и определяющих хромосомную и геномную стабильность. Теломерный комплекс состоит из каталитической субъединицы обратной транскриптазы теломеразы (TERT), РНК-компонента транскриптазы (hTR) и дискерина (DKC1). Было показано, что TERT является интегральным белком, определяющим активность теломеразного комплекса. Ген TERT располагается на 5 хромосоме и включает 16 экзонов. Промотор гена TERT имеет размер в 330 нуклеотидов и содержит GC-насыщенные регионы, содержащие места связывания для нескольких факторов транскрипции. Более чем 20 лет назад было показан феномен повышенной активности теломераз при различных опухолевых процессах, однако мутации в кодирующей области гена TERT встречаются крайне редко. Относительно недавно было обнаружено, мутации, вызывающие гиперактивность теломераз при различных типах опухолей, располагаются в промоторной зоне и представляют собой две точечные мутации–124G>A (C228T) и –146G>A(C250T), которые формируют новые точки связывания (GGAA) для транскрипционных факторов ETS, что также проявляется повышенной экспрессией теломераз. Клетки и ткани щитовидной железы редко подвергаются обновлению во взрослом возрасте, что объясняет низкую активность теломераз в норме. При РЩЖ активация теломеразы встречается реже, чем в других видах злокачественных новообразований и тесно связано с уровнем дифференцировки рака. Распространённость мутации –124G>A (C228T) и –146G>A (C250T) при РЩЖ, в соответствие с последними работами, сильно варьирует и составляет 7-50%. Мутации в промоторе TERT-гена ассоциированы с более тяжелым и агрессивным течением РЩЖ, особенно при обнаружении данных аберраций при ПК.
Заключение
На сегодняшний день в соответствии с международными и отечественными рекомендациями молекулярно-генетическое обследование является одним из инструментов для подтверждения наличия злокачественного новообразования при ТАБ узлов щитовидной железы. Кроме этого, ряд молекулярно- генетических маркеров может предсказывать более тяжелое течение заболевания.
| Молекулярное тестирование | Препараты, одобренные на территории РФ, а также используемые в рамках клинических исследований (*) |
| BRAF | ТКИ BRAF/MEK: дабрафениб+траметиниб* |
Зеленый — чувствительность к терапии.
| Ген | Краткая информация |
| Мутация V600E в гене BRAF | • Распространенность мутации V600E в гене BRAF при папиллярной карциноме составляет 40-50%, при низкодифференцированной карциноме- 20-40%, при анапластической карциноме 30-40%. • Наличие мутации V600E в гене BRAF обуславливает риск обнаружения злокачественного процесса в >95% случаев (ATA, 2015). • Мутация V600E увеличивает вероятность обнаружения агрессивных клинико-морфологических характеристик. • Наличие мутации V600E в гене BRAF при папиллярном РЩЖ, размером более 1 см, определяет высокий риск рецидива после начального лечения (>40%). Показания: Рекомендовано проведение генетического тестирования цитологического материала промежуточного риска злокачественности по Bethesda (III-V). Рекомендовано всем пациентам с высокодифференцированным раком щитовидной железы для послеоперационной стратификации риска рецидива для определения послеоперационной тактики ведения пациентов. |
| Мутации С228Т и С250Т в промотерном регионе гена TERT | • Наличие мутации в промотерном регионе гена TERT обуславливает риск обнаружения злокачественного процесса в >95% случаев (ATA, 2015). • Рядом авторов показано, что мутация в промотерном регионе гена TERT ассоциирована с риском рецидива заболевания и смертностью, а также взаимосвязана с агрессивным гистологическим фенотипом (ETA, 2017). • Наличие мутации С228Т и С250Т в гене TERT изолированно либо в комбинации с мутацией V600E в гене BRAF при папиллярном РЩЖ, размером более 1 см, определяет высокий риск рецидива после начального лечения (>40%) (ATA, 2015). Показания: Рекомендовано проведение генетического тестирования цитологического материала промежуточного риска злокачественности по Bethesda (III-V). Рекомендовано всем пациентам с высокодифференцированным раком щитовидной железы для послеоперационной стратификации риска рецидива для определения послеоперационной тактики ведения пациентов. |
| Мутации в генах RAS | • Распространенность мутаций в генах семейства RAS при фолликулярном варианте папиллярной карциномы составляет 30-45%, при фолликулярной карциноме- ~45%, при анапластической карциноме 10-20%. • Наличие мутации в генах семейства RAS (KRAS, NRAS, HRAS) обуславливает риск обнаружения злокачественного процесса в цитологическом материале категории Bethesda III, IV в 84% случаев (ATA, 2015). • Цитологические образцы категории Bethesda III, IV с наличием мутаций в генах семейства RAS могут рассматриваться в аналогичной категории риска, что и цитологический материал с подозрением на рак (Bethesda V) (ATA, 2015). Показания: Рекомендовано проведение генетического тестирования цитологического материала промежуточного риска злокачественности по Bethesda (III-V). |
| Транслокация PAX8/PPARG | • Распространенность транслокации PAX8/PPARG при низкодифференцированной карциноме щитовидной железы составляет 3,6% (MSKCC,2016). • Наличие транслокации PAX8/PPARG обуславливает риск обнаружения злокачественного процесса в цитологическом материале категории Bethesda III-V в >95% случаев (ATA, 2015). Показания: Рекомендовано проведение генетического тестирования цитологического материала промежуточного риска злокачественности по Bethesda (III-V). |
Список литературы
- Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016. doi:10.1089/thy.2015.0020.
- Sahli ZT, Smith PW, Umbricht CB, Zeiger MA. Preoperative Molecular Markers in Thyroid Nodules. Front Endocrinol (Lausanne). 2018;9:179. Published 2018 Apr 18. doi:10.3389/fendo.2018.00179
- Penna G, C, Vaisman F, Vaisman M, Sobrinho-Simões M, Soares P: Molecular Markers Involved in Tumorigenesis of Thyroid Carcinoma: Focus on Aggressive Histotypes. Cytogenet Genome Res 2016;150:194-207. doi: 10.1159/000456576
- Eszlinger M, Lau L, Ghaznavi S, Symonds C, Chandarana SP, Khalil M, et al. Molecular profiling of thyroid nodule fine-needle aspiration cytology. Nat Rev Endocrinol 2017. doi:10.1038/nrendo.2017.24.
- Roth MY, Witt RL, Steward DL. Molecular testing for thyroid nodules: Review and current state. Cancer 2018. doi:10.1002/cncr.30708.
- DeVita, Vincent T., Jr., Theodore S. Lawrence, and Steven A. Rosenberg. Devita, Hellman, and Rosenberg’s Cancer: Principles & Practice of Oncology. 11th edition. Philadelphia: Wolters Kluwer, 2019.
- Nishino M, Nikiforova M. Update on molecular testing for cytologically indeterminate thyroid nodules. Arch. Pathol. Lab. Med., 2018. doi:10.5858/arpa.2017-0174-RA.