Немелкоклеточный рак легкого

Рак легкого по сей день остается наиболее распространенной причиной смертности от злокачественных новообразований: так в 2018 заболевание привело к смерти 1,8 миллионов человек, что составило около 20% от общей смертности от злокачественных новообразований. Высокая социальная значимость заболевания привела к значительному продвижению в понимании молекулярной биологии опухоли, в частности, наиболее распространенного типа — немелкоклеточного рака легкого (НМРЛ). НМРЛ составляет до 85% всех случаев рака легкого.
Патогенез НМРЛ представляет собой сложный многоступенчатый процесс, включающий в себя генетические аберрации, активацию сигнальных путей роста клеток и ингибирование их супрессоров. Мутации в генах KRAS и BRAF были одним из первых генетических маркеров, обнаруженных в опухолевых клетках аденокарциномы легкого. Идентификация активирующих мутаций в гене рецептора эпидермального фактора роста (EGFR) привела к разработке и клиническому использованию таргетных препаратов, специфически ингибирующих повышенную тирозинкиназную активность мутантных белковых молекул. В последующем, список активирующих мутаций при раке легкого был значительно расширен: мутации были обнаружены в генах HER2 (также известный как ERBB2), рецептор фактора роста фибробластов 1 (FGFR1) и FGFR2, а также онкогенах анапластической лимфомакиназы (ALK), тирозинкиназы рецептора ROS1, тирозин-протеинкиназы Met, рецептора нейротрофической тирозинкиназы типа 1 (NTRK1) и RET. Для многих из перечисленных генов также существуют или разрабатываются препараты, выборочно блокирующие поврежденные пути активации вторичных посредников. Для плоскоклеточного рака легкого было идентифицировано значительно меньше количество генов, мутации в которых вызывают злокачественный рост. Нужно также отметить, что спектр генов, повреждающихся при аденокарциноме и плоскоклеточном раке легкого, значительно отличается. Так, для плоскоклеточного рака легкого характерны мутации в таких генах, как дискоидин домен содержащий рецептор 2 (DDR2), FGFR1, FGFR2, FGFR3 и генах, участвующих в экспрессии белков сигнального пути PI3K. Многие из этих мутаций, за исключением мутаций в генах, участвующих в пути PI3K, являются драйверными мутациями. Исследования с помощью секвенирования нового поколения продемонстрировали невероятную сложность соматических изменений в НМРЛ, которые затрагивают помимо белковых киназ модификаторы эпигенома, факторы транскрипции, факторы сплайсинга и гены, участвующие в клеточном иммунитете.
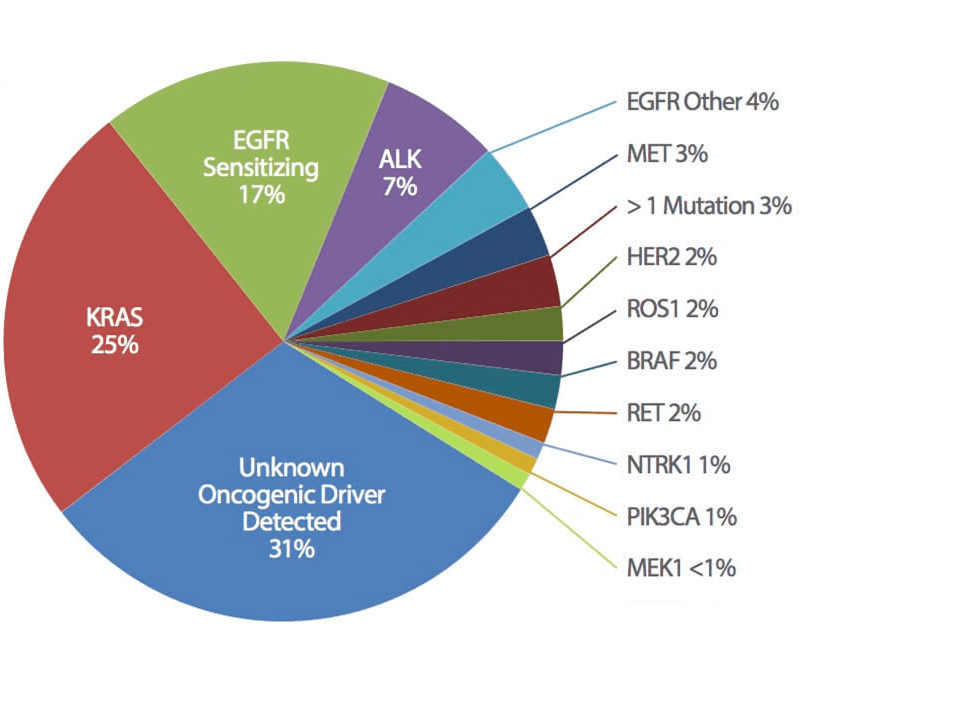
Рак легкого характеризуется значительной генетической гетерогенностью при относительно небольшом количестве клинически значимых мутаций, которые встречаются в большинстве случаев. При этом, НМРЛ имеет высокую мутационную нагрузку. Медиана частоты соматических мутаций составляет десять мутаций на мегабазу кодирующей последовательности ДНК. Стоит отметить, что аденокарцинома у некурящих содержала в 5-6 раз меньше мутаций. Учитывая относительно большое количество мутаций на одну опухоль, вероятно, будут обнаружены более важные мутации для НМРЛ, поскольку число анализируемых опухолей увеличивается.
Геномная и хромосомная нестабильность способствует гетерогенности в солидных опухолях. В нормальных легочных клетках геном реплицируется с высокой точностью, а мутации редко допускаются, что объясняется строгими механизмами внутренней проверки, такими как восстановление оснований и нуклеотидов, мисмэтч-репарация, поддержание длины теломер и репарация разрывов двуспиральной ДНК. Неисправность механизмов контроля значительно увеличивает частоту мутаций и значительно ускоряет процесс клональной эволюции, приводя к высокой клональной и генетической гетерогенности опухолей. Показано, что хромосомная нестабильность коррелирует с более короткой выживаемостью у пациентов с НМРЛ, возможно, потому что эти нестабильные опухолевые клетки проявляют более высокую множественную лекарственную устойчивость по сравнению со стабильными клетками.
Одним из главных факторов, способствующих генетическому разнообразию НМРЛ, являются клетки-источники, из которых возникают генетические подтипы НМРЛ. До сих остается неясным, возникает ли опухоль из одной аберрантной клетки с драйверной мутацией, возникшей в благоприятном микроокружении, или же существует возможность появления одной и той же генетической поломки в нескольких типах клеток, что приводит к развитию опухолевого клона. Существует вероятность, что генетические особенности клеток-предшественников имеют прямое влияние на генотип и фенотип зрелой опухоли. Это может быть следствием уникального профиля экспрессии генов в клетках-предшественниках, различий их гистологического типа или же микроокружения клеток. Считается, что источником опухолевых клеток-предшественников рака являются стволовые и другие тотипотентные клеточные линии, так как только они имеют достаточное время существования для аккумулирования большого количества генетических аберраций, которые требуются для активации туморогенеза. Более того, для стволовых клеток не требуется значительного эпигенетического репрограммирования, что объясняется повышенными возможностями самообновления.
Идентификация биологически значимых генетических альтераций, а также определение их клональности или субклональности при НМРЛ дает возможность разработки новых терапевтических подходов. Понимание молекулярного патогенеза НМРЛ значительно расширилось с внедрением в практику методов параллельного массового секвенирования. В соответствии с последними полученными данными полноэкзомного секвенирования опухолевого материала рака легкого, новообразования данного типа обладают значительной генетической гетерогенностью и разнообразием.
По результатам последних исследований было обнаружено более 700 раннее неизвестных генетических соматических изменений. Важной задачей, которая остается, является понимание того, какие из этих многих мутаций важны для канцерогенеза и/или ответа на лечение и какие являются следствием онкогенного процесса. Таким образом, геномные профили подчеркивают гетерогенность генома НМРЛ и дают вполне убедительное объяснение высоко гетерогенных ответов на лечение, которые наблюдаются в клинической практике. Систематизируя большой набор мутаций для каждого пациента, может быть достигнута более точная оценка общих эффектов генотипа на ответ терапии, что в конечном счете поможет максимально персонализировать подходы к терапии.
EGFR
EGFR, рецептор эпидермального фактора роста, относится к семейству тирозинкиназных рецепторов, которое также включает человеческий рецептор эпидермального фактора роста 2 (HER2, также известный как ERBB2), HER3 (ERBB3) и HER4 (ERBB4). Данный рецептор содержит четыре внеклеточных домена, трансмембранный домен, тирозинкиназный домен и С-концевой хвост. Связывание активирующих лигандов приводит к димеризации EGFR и транс-фосфорилированию остатков тирозина в С-концевом хвосте с активацией нижележащих сигнальных путей, таких как PI3K/AKT/mTOR, RAS/RAF/MAPK и JAK/STAT. Данные пути вовлечены в регуляцию клеточной пролиферации, выживании, инвазии и ангиогенезе.
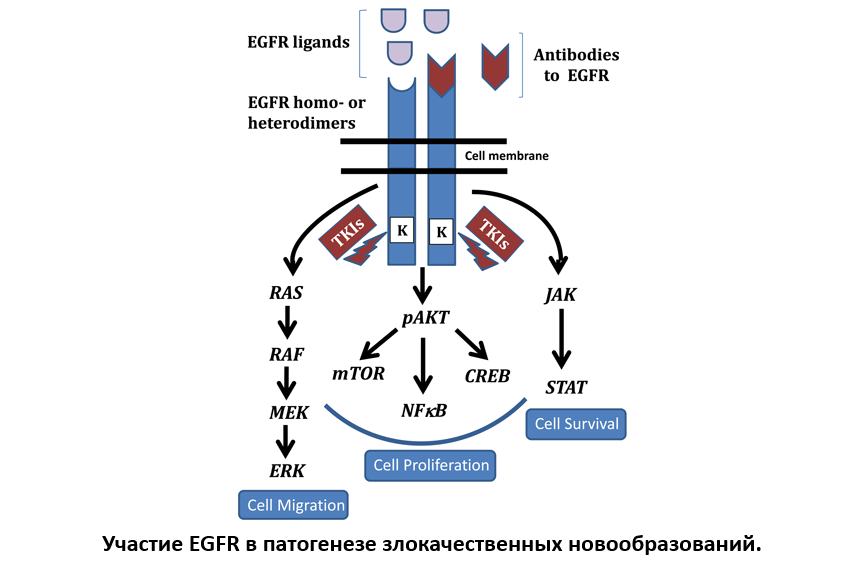
Мутации в гене EGFR составляют 10-20% случаев аденокарциномы. Данные аберрации затрагивают АТФ-связывающий карман тирозинкиназный домен, что приводят к конститутивной активации и, как следствие, независимости от лигандов. Наиболее распространенными мутациями в гене EGFR, ассоциированные с чувствительностью к тирозинкиназным ингибиторам (ТКИ) EGFR, являются делеции в 19 экзоне и миссенс-мутацию в 21 экзоне- L858R. ТКИ EGFR первого поколения, такие как гефитиниб и эрлотиниб, имеют более высокие показатели частоты объективного ответа и безрецидивной выживаемости по сравнению с цитотоксической терапией у ранее нелеченых пациентов с наличием активирующих мутаций в гене EGFR. В отличие от ТКИ EGFR первого поколения, которые являются обратимыми ингибиторами и нацелены только на EGFR, ингибиторы второго поколения, включая афатиниб и дакомитиниб, являются необратимыми ингибиторами, которые также нацелены на HER2 и HER4. Как афатиниб, так и дакомитиниб имеет выше показатели безрецидивной выживаемости по сравнению с гефитинибом. Афатиниб также продемонстрировал значительное увеличение медианы общей выживаемости по сравнению с цитотоксической терапией на основе платины у пациентов с делецией в 19 экзоне, но не у пациентов с мутацией L858R. Причиной такой особенности является различия в конформационных изменениях АТФ-связывающего кармана, а также паттерна аутофосфорилирования, индуцируемого каждой мутацией.
Наиболее распространенной причиной приобретенной устойчивости к первому поколению ТКИ EGFR является появление второй мутации в 20 экзоне гена EGFR, приводящая к замене треонина на метионин в 790 кодоне (T790M). Данная мутация приводит к повышению аффинности тирозинкиназного домена к АТФ. Другими механизмами резистентности являются амплификация гена HER2, мутации в генах MET, BRAF или PIK3CA, а также трансформация в мелкоклеточный рак легкого. Это показывает необходимость повторного молекулярного профилирования при прогрессии заболевания для определения дальнейшей тактики ведения пациента. ТКИ EGFR третьего поколения являются селективными ингибиторами аберрантного EGFR, обусловленного наличием активирующих мутаций, а также мутацией T790M. При этом, ингибиторы третьего поколения не затрагивают EGFR дикого типа. Эти препараты ковалентно связываются с цистеином в кодоне 797, что позволяет преодолеть повышенную аффинность домена к АТФ в результате мутации T790M. Осимертиниб, ТКИ EKFR третьего поколения, эффективен у пациентов с мутацией T790M, выявленной в результате прогрессирования при использовании ТКИ EGFR первого поколения. Данный препарат продемонстрировал более высокие показатели частоты объективного ответа и безрецидивной выживаемости по сравнению с цитотоксической терапией на основе платины. В рандомизированном исследовании, в котором сравнивали осимертиниб с эрлотинибом или гефитинибом у ранее нелеченых пациентов с метастатическим НМРЛ с наличием делеции в 19 экзоне либо мутации L858R, осимертиниб продемонстрировал более высокие показатели безрецидивной выживаемости (18,9 против 10,2 месяцев, соответственно, p<0,001) и медианы общей выживаемости (38,6 против 31,8 месяцев, соответственно, p=0,046). Таким образом, осимертиниб может быть использован в качестве первой линии терапии у ранее нелеченых пациентов с наличием активирующих мутаций в гене EGFR (NCCN, 2020).
ALK
ALK представляет собой трансмембранный тирозинкиназный рецептор, функция которой в норме у человека изучена не до конца. ALK был впервые обнаружен в 1997 году как химерный белок, возникший в результате транслокации t (2; 5) (p23; q35) при анапластической крупноклеточной лимфоме. В 2007 году при НМРД была обнаружена другая транслокация ALK. При ALK транслокациях наиболее распространенным партнером слияния является ген EML4 (EML4-ALK). Продукт слияния гена ALK конститутивной активностью в цитоплазме вне зависимости от лиганда, что приводит к активации сигнальных путей RAS/RAF/MEK, PI3K/AKT/mTOR и JAK/STAT. Транслокации ALK наблюдаются в 2-5% случаев НМРЛ, преимущественно у молодых некурящих пациентов.
Кризотиниб обладает активностью в отношении ALK-позитивных опухолей и является конкурентным ингибитором тирозинкиназ ALK, MET и ROS1. Данный препарат имеет более высокие показатели частоты объективного ответа и безрецидивной выживаемости по сравнению с цитотоксическими препаратами как у принимавших ранее терапию, так и ранее нелеченых пациентов с наличием транслокаций гена ALK. Большинство пациентов, ранее получавших кризотиниб, имели ответ при терапии ингибиторами ALK второго поколения, включая церитиниб, алектиниб и бригатиниб. Церитиниб также имеет более высокие показатели частоты объективного ответа и безрецидивной выживаемости по сравнению с цитотоксической терапией у пациентов с ALK-позитивным НМРЛ. В ходе двух рандомизированных исследований было показано увеличение показателей частоты объективного ответа и безрецидивной выживаемости при использовании алектиниба по сравнению с кризотинибом у ранее нелеченых ALK-позитивных пациентов. Таким образом, алектиниб является предпочтительным препаратом в качестве первой линии терапии у данной группы пациентов.
KRAS
KRAS представляет собой протоонкоген из семейства белков RAS и кодирует G-белок, активно участвующей в регуляции и контроле клеточной пролиферации, дифференцировки и выживании. Белок Ras неактивен в нормальных не пролиферирующих клетках, но вступает в работу при связывании с гуанозинтрифосфатом, что приводит к реактивации рецепторов факторов роста. Активированные комплекс Ras-ГТФ связывается и активирует ряд вторичных посредников передачи клеточного сигнала, такие как митоген активирующую протеин киназу (MAPK), белки путей RAS/RAF/MEK/MAPK и PI3-K [PI3K/AKT/mTOR]. KRAS играет основную роль в контроле передачи сигнала от ряда основных рецепторов факторов роста, в том числе EGFR. Активирующие мутации в гене KRAS изменяют ГТФ-азную активность белка и блокируют возможность его деактивации, что ведет к гиперактивации ряда внутриклеточных путей передачи сигнала, контролирующих рост и пролиферацию клеток.
Мутации в RAS/RAF/MEK/MAPK-сигнальном пути играют основную роль в развитии рака легкого.

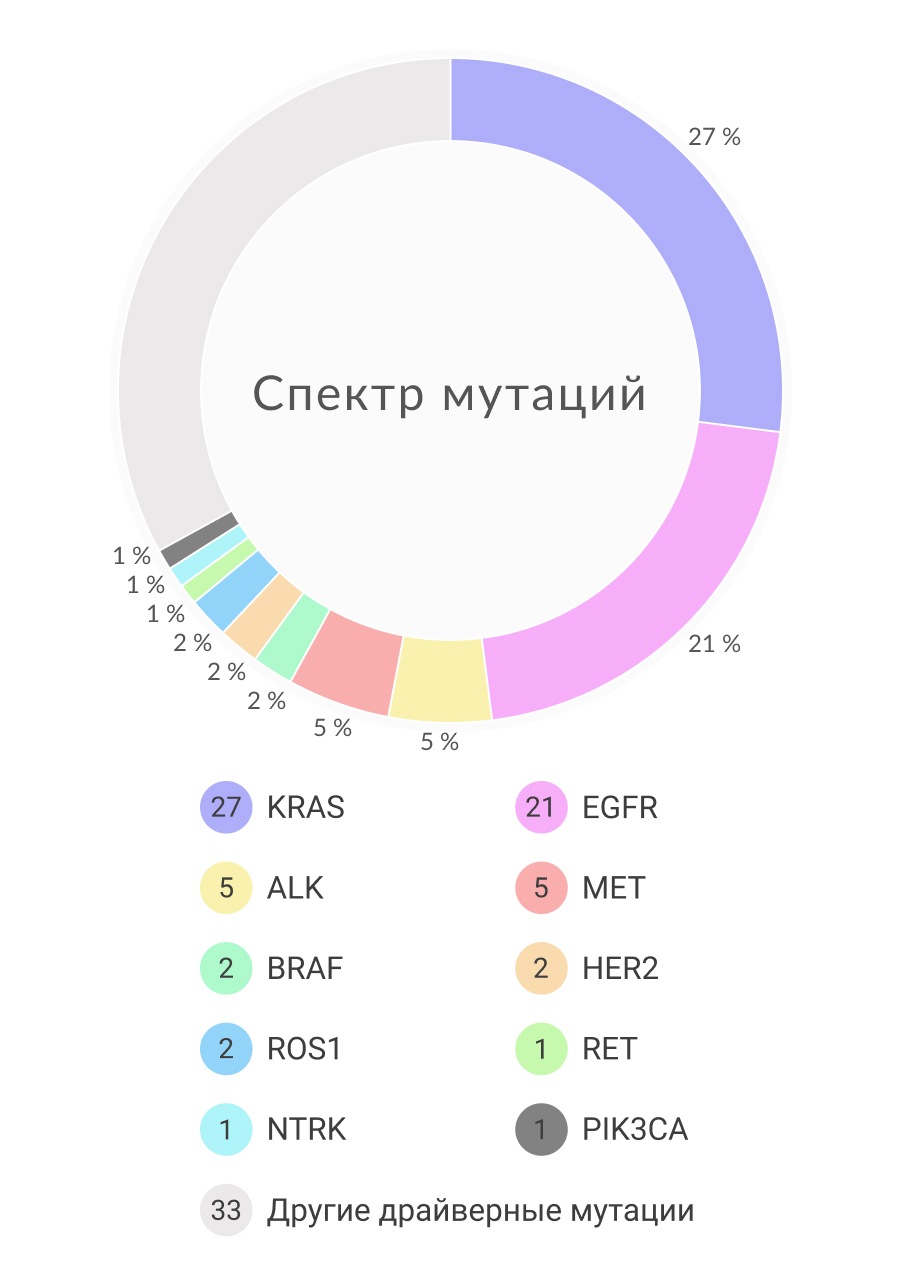
Чаще всего аберрации наблюдались в гене KRAS. Активирующие мутации в онкогене KRAS представляют собой наиболее частую генетическую аберрацию при аденокарциноме легкого, встречающуюся приблизительно в 25% случаев НМРЛ. Высокая вариация распространённости мутаций в гене KRAS связана с этническими, половыми различиями, а также факторами влияния внешней среды исследуемых выборок пациентов. Так, мутации в данном гене более распространены в европейской популяции, чем у пациентов из Азии. У пациентов с аденокарциномой легкого, которые никогда не курили, распространённость аберраций в гене KRAS составила 0-15%. Самой частой мутацией в гене KRAS при аденокарциноме лёгкого является аберрации в 12 кодоне, реже встречаются мутации в кодоне 13 и 61.
Учитывая, что аберрации в гене KRAS представляют собой драйверные мутации, они редко встречаются совместно с изменениями в гене EGFR. Было показано, что пациенты с KRAS-ассоциированным раком легкого резистентны к терапии ингибиторами EGFR, так как мутации в гене KRAS ведет к активации вторичных посредников, находящихся выше в цепочке сигнального пути, чем белки гена EGFR. Существуют данные о том, что различные генетические аберрации в гене KRAS имеют разное клиническое значение. В соответствии с исследованием BATTLE, альтерации G12C и G12V в гене KRAS предсказывают более быстрое прогрессирование заболевания у пациентов по сравнению с другими типами KRAS-мутаций или отсутствием генетических изменений в этом гене. Более того, различные мутации ведут к активации разных путей передачи сигнала, что объясняется разницей в конформации мутантного белка и, как следствие, разницей специфичности к нижестоящим медиаторам передачи сигнала. Это еще раз подчеркивает важность учета генетической составляющей рака легкого при разработке новых таргетных лекарственных средств.
BRAF
BRAF представляет собой серин/треониновую киназы, которая задействована в сигнальном пути MAPK, участвующий в регуляции клеточной выживаемости и пролиферации. Активирующие мутации в BRAF имеют высокую распространённость при меланоме, но они редко встречаются при НМРЛ. Спектр мутаций при НМРЛ значительно отличается от такого при меланоме и колоректальном раке. При НМРЛ мутация V600E, активирующая киназный домен белка, встречается реже, чем при других видах опухолей. При аденокарциноме легкого мутация V600E обнаруживается в 50% случаев от всех BRAF мутаций. Другими частыми аберрациями являются G469A и D594G. Некоторые BRAF-мутации при НМРЛ появляются в киназном домене (V600E, D594G и L596R), другие же встречаются в участке гена, кодирующего G-петлю домена активации белка (G465V и G468A). Так как белки генов BRAF и KRAS представляют собой компоненты единого сигнального пути, в основе которого стоит EGFR, мутации в одном из генов практически полностью исключают аберрации в двух других генах. Мутации в гене BRAF в основном встречаются при аденокарциноме лёгкого. Согласно рекомендациям RUSSCO при наличии активирующей мутации BRAF V600E у пациентов с метастатическим НМРЛ показано применение комбинации BRAF- и MEK-ингибиторов: дабрафениб+траметиниб.
ROS1
Онкоген ROS1 является тирозинкиназным рецептором. Транслокации данного гена являются драйверным событием в 1-2% случаев НМРЛ. ROS1 большое количество партнеров слияния, однако наиболее распространенным из них является CD74. ROS1-позитивный НМРЛ имеет следующие особенности: гистологически представлен аденокарциномой, встречается у молодых никогда не куривших пациентов. Аберрантный ROS1 чувствителен как к терапии ингибитором тропомиозин-рецепторной киназы, энтректинибу, так и к кризотинибу благодаря высокой степени гомологии между тирозинкиназными доменами ALK и ROS1. И энтректиниб, и кризотиниб одобрены FDA для пациентов с транслокацией ROS1, включая тех, кто получил химиотерапию, и тех, кто ранее не получал лечение. Однако, энтректиниб предпочтителен у пациентов с наличием метастазов в головной мозг ввиду эффективного проникновения препарата через гематоэнцефалический барьер.
MET
Протоонкоген MET располагается на хромосоме 7q21-q31 и кодирует мембранный тирозинкиназный рецептор, который также известен, как рецептор фактора роста гепатоцитов. При связывании лиганда с данным рецептором происходит гомодимеризация рецептора, активация киназы и индуцирование RAS/RAF/MEK/MAPK, PI3K/AKT и c-SRC сигнальных путей. При НМРЛ происходит амплификация гена MET в 1-7% случаев. Увеличение количества копий гена MET приводит к гиперэкспрессии белка и активации сигнальных путей. Онкогенная активность МЕТ была продемонстрирована in vitro: амплификация гена приводила к конституциональному фосфорилированию рецептора, активации PI3K/AKT-сигнального пути. Амплификация гена МЕТ представляет собой одну из причин вторичной резистентности к ингибиторам тирозинкиназ, которая наблюдается в 20% случаев резистентности к терапии ТКИ EGFR. В этом случае амплификация МЕТ поддерживает активацию PI3K/AKT-сигнального пути в обход блокировки EGFR.
HER2
Человеческий рецептор эпидермального фактора роста 2 (HER2 [ERBB2]) представляет собой тирозинкиназный рецептор семейства EGFR. Мутации в HER2 обнаруживаются примерно в 1–3% случаев НМРЛ. К активирующим мутациям гена HER2 относят инсерции в 20 экзоне размером 3-12 нуклеотидов, но также наблюдались точечные мутации в 20 экзоне. Эти опухоли являются преимущественно аденокарциномами, более распространены среди тех, кто никогда не курил, и большинство этих пациентов — женщины.
NTRK
Соматические транслокации, затрагивающие гены нейротрофной рецепторной тирозинкиназы (NTRK1-3), также являются онкогенным драйверным событием и наблюдаются в менее 1% случаев НМРЛ. Также для транслокаций NTRK характерно большое количество партнеров слияния. В настоящее время не показано клинико-морфологических особенностей NTRK-позитивных опухолей. Для лечения пациентов с данной транслокацией FDA одобрены два препарата: ларотректиниб и энтректиниб.
RET
Ген RET располагается в локусе 10q11. и кодирует тирозинкиназный рецептор, который участвует в развитии нейрональной трубки. Изменения в гене RET ассоциированы с раком щитовидной железы, но было показано, что активация RET происходит в некоторых случаях рака легкого, путем транслокации гена. Транслокация участка гена RET, кодирующего киназный домен, происходит к гену KIF5B, который также располагается на 10 хромосоме и кодирует белок, участвующий в транспорте органелл. KIF5B-RET была обнаружена в 1-2% случаев аденокарциномы легкого, при этом наличие данной аберрации исключает возможность мутаций в генах EGFR, KRAS и ALK.
Заключение
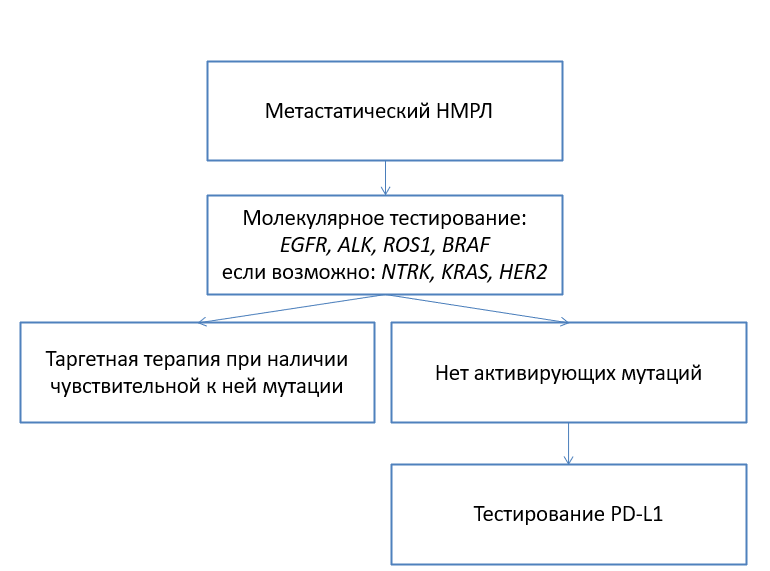
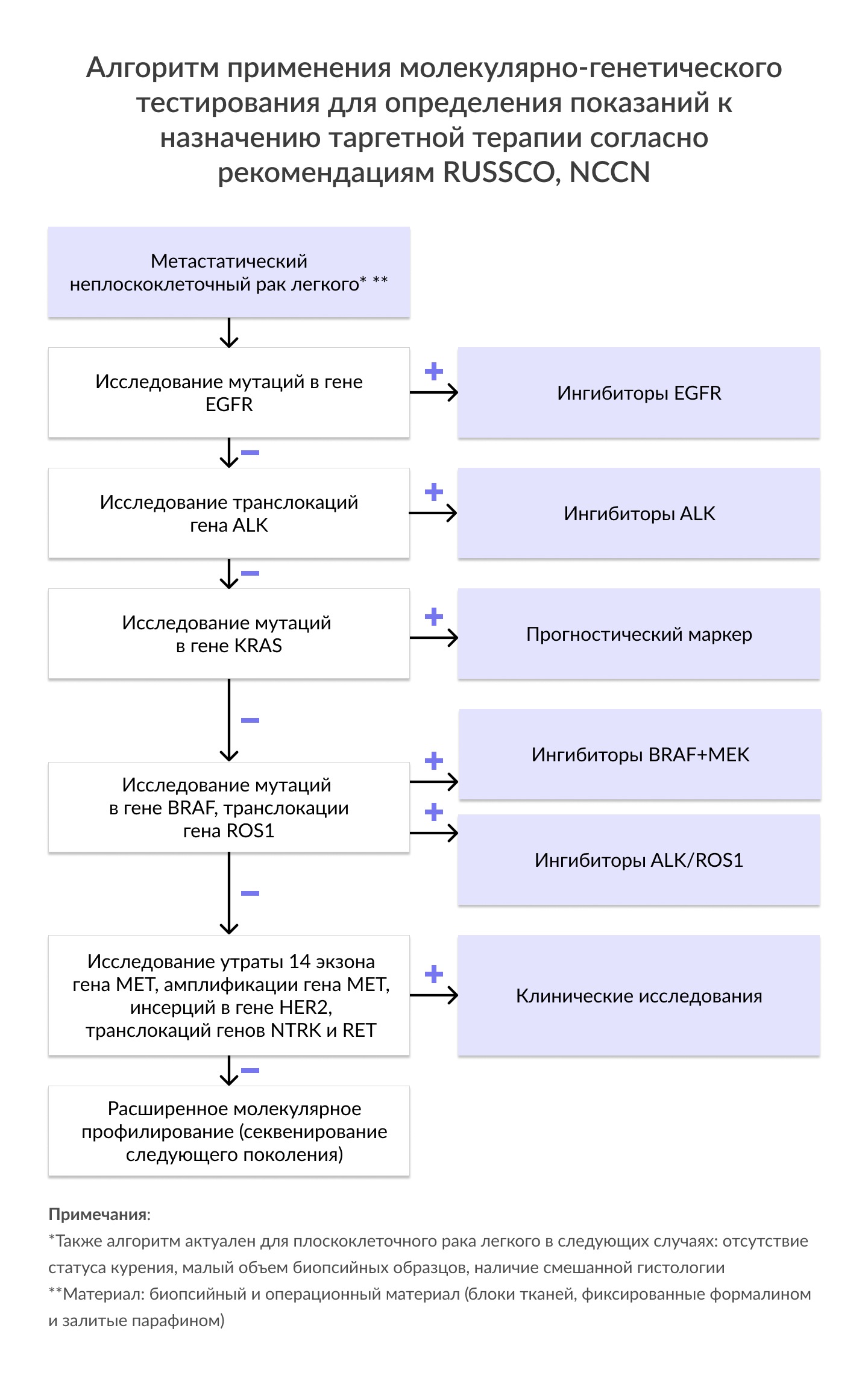
Молекулярно-генетическое исследование опухолевого материала рака легкого является обязательным этапом обследования пациентов в связи с возможностью проведения таргетной терапии носителей специфических мутаций.
| Молекулярное тестирование | Препараты, одобренные на территории РФ, а также используемые в рамках клинических исследований/программы расширенного доступа (*) |
| EGFR | Тирозинкиназные ингибиторы (ТКИ) EGFR: гефитиниб, эрлотиниб, афатиниб, осимертиниб |
| KRAS | Соторасиб* (только KRAS G12C), ТКИ EGFR |
| ALK | Алектиниб, церитиниб, кризотиниб, бригатиниб*, лорлатиниб* |
| ROS1 | Кризотиниб, энтректиниб* |
| BRAF | ТКИ BRAF/MEK: дабрафениб+траметиниб |
| NTRK | Капматиниб*, тепотиниб*, кризотиниб |
| MET | Капматиниб*, кризотиниб |
| HER2 | Адо-трастузумаб эмтанзин*,фам-трастузумаб дерукстекан- nxki* |
Зеленый — чувствительность к терапии, Красный — резистентность к терапии.
| Ген | Краткая информация |
| Мутации в гене EGFR | • Мутации в 18-21 экзонах гена EGFR встречаются у 10-20% пациентов с аденокарциномой легкого (ESMO, 2019). |
| Мутации в гене KRAS | • Точечные мутации в гене KRAS встречаются приблизительно в 25% случаев. • Аберрации в данном гене ассоциированы с низкой выживаемостью. • Альтерации в гене KRAS также связаны с резистентностью к таргетной терапии. • Наличие мутаций в данном гене не оказывает влияния на эффективность химиотерапии. • Отсутствие мутаций в гене KRAS значительно увеличивает вероятность обнаружения чувствительных к таргетной терапии аберраций в генах EGFR, ALK, ROS1 у пациентов с немелкоклеточным раком легкого, так как альтерации в перечисленных генах обычно не перекрываются. Показания: Рекомендовано пациентам с метастатическим НМРЛ для определения возможных вариантов таргетной терапии. |
| Мутация V600E в гене BRAF | • Распространенность мутации V600E в гене BRAF- около 2%. • Мутации в гене BRAF в основном встречаются при аденокарциноме лёгкого и чаще всего ассоциированы с наличием статуса курения. Показания: Рекомендовано всем пациентам с метастатическим НМРЛ для определения показаний к назначению комбинированной таргетной терапии тирозинкиназными ингибиторами BRAF/MEK. |
| Транслокация ALK | • Распространенность перестроек гена ALK составляет около 5% (NCCN, 2020). • Наиболее частым типом транслокаций является EML4-ALK (NCCN, 2020). • Вариант 1 и 3a/b являются наиболее распространенными вариантами перестройки EML4-ALK, которые обнаруживаются в 33% и 29% случаев EML4-ALK-позитивного НМРЛ, соответственно (Sabir S., 2017). Показания: Рекомендовано проведение ALK-тестирования у пациентов с метастатическим НМРЛ для определения возможности применения таргетной терапии тирозинкиназными ингибиторами ALK. |
| Транслокация ROS1 | • Распространенность перестроек гена ROS1 составляет около 1-4% (ESMO, 2019). • Наиболее частым типом транслокаций является CD74-ROS1, который составляет 40-45% случаев ROS1-позитивных опухолей. Показания: Рекомендовано проведение ROS1-тестирования у пациентов с метастатическим НМРЛ для определения возможности применения таргетной терапии тирозинкиназными ингибиторами ALK/ROS1 (кризотиниб). |
| Экспрессия мРНК PD-L1 | • Терапия ингибиторами контрольных точек иммунитета оказывается неэффективным во всех случаях низкой экспрессии PD-L1 (<1%). Показание: Рекомендовано пациентам с метастатическим НМРЛ для определения показаний к проведению иммунотерапии. |
| Утрата 14 экзона гена MET | • Утрата 14 экзона гена MET наблюдается в 3-4% случаев аденокарцином легкого. • Данная аберрация чаще встречается у некурящих женщин пожилого возраста. • У пациентов с утратой 14 экзона гена MET наблюдаются умеренные показатели частоты ответа (16%) при проведении иммунотерапии, даже несмотря на наличие высокого уровня экспрессии PD-L1. • FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов) был одобрен капматиниб (возможно использование в РФ в рамках клинического исследования) для лечения пациентов с утратой 14 экзона гена MET. • Также в рамках клинического исследования возможно применение кризотиниба для лечения пациентов с метастатическим НМРЛ и наличием данной аберрации. Показания: Рекомендовано пациентам с метастатическим НМРЛ для определения показаний к проведению таргетной терапии (капматинибом или кризотинибом) в рамках клинического исследования. |
| Амплификация гена MET | • Распространенность амплификации гена MET составляет 1-5% случаев НМРЛ. • Данная аберрация является одной из причин приобретенной резистентности к тирозинкиназным ингибиторам 1 и 2 поколений (эрлотиниб, гефитиниб, афатиниб). Так, амплификация гена MET отмечается в 15-22% случаев приобретенной резистености. • В рамках клинического исследования возможно применение кризотиниба для лечения пациентов с метастатическим НМРЛ и наличием данной аберрации. Показания: Рекомендовано пациентам с метастатическим НМРЛ для определения показаний к проведению таргетной терапии (кризотинибом) в рамках клинического исследования. |
Список литературы
- DeVita, Vincent T., Jr., Theodore S. Lawrence, and Steven A. Rosenberg. Devita, Hellman, and Rosenberg’s Cancer: Principles & Practice of Oncology. 11th edition. Philadelphia: Wolters Kluwer, 2019.
- Ettinger DS, Wood DE, Aggarwal C, Aisner DL, Akerley W, Bauman JR, et al. NCCN Guidelines Insights: Non–Small Cell Lung Cancer, Version 1.2020. J Natl Compr Cancer Netw J Natl Compr Canc Netw 2019;17:1464–72. doi:10.6004/jnccn.2019.0059.
- Lavdovskaia E.D., Iyevleva A.G., Sokolenko A.P., Mitiushkina N.V., Preobrazhenskaya E.V., Tiurin V.I. et al. EGFR T790M Mutation in TKI-Naive Clinical Samples: Frequency, Tissue Mosaicism, Predictive Value and Awareness on Artifacts. Oncol Res Treat. 2018.
- Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann Oncol 2018. doi:10.1093/annonc/mdy275.
- Del Re M., Rofi E., Restante G., Crucitta S., Arrigoni E., Fogli S. et al. Implications of KRAS mutations in acquired resistance to treatment in NSCLC. Oncotarget. 2018.
- Lim S.M., Syn N.L., Cho B.C., Soo R.A. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat Rev. 2018.
- Arora R., Krishnan V. Selective Targeting of the L858R Mutation (EGFR) in Non-Small Cell Lung Cancer: A Mechanism for Advancing Targeted Chemotherapy. Front Oncol. 2017; 7.